One in 2,500 people (i.e. one birth every four days) is born with cystic fibrosis, making it the most common incurable, life-limiting disorder in Australia (Healthdirect 2023; Better Health Channel 2024).
What is Cystic Fibrosis?
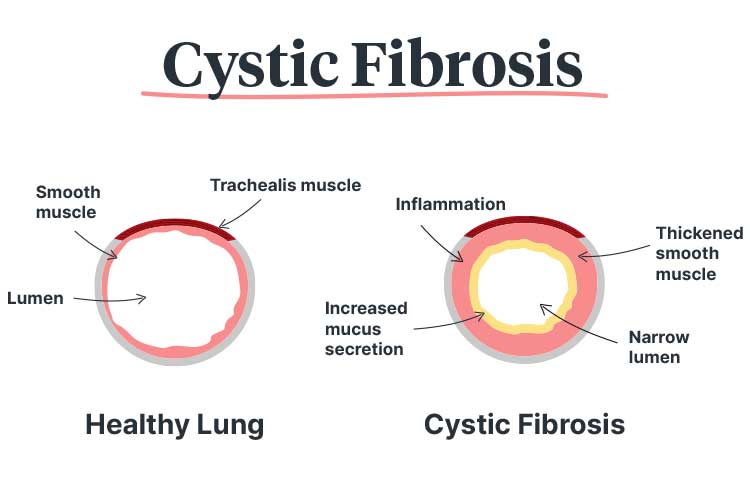
Cystic fibrosis (CF) is a life-limiting genetic condition in which abnormally thick and sticky mucus is produced, causing complications that predominantly affect the respiratory, digestive and reproductive systems (Better Health Channel 2024).
CF is incurable and shortens life expectancy; however, this has rapidly improved in recent years due to improvements in care. Now, the average life expectancy is 49 years for people born between 2005 and 2009 and 56 years for people born between 2016 and 2020 (Monash University 2023).
How Does Cystic Fibrosis Affect the Body?
Cystic fibrosis causes a build-up of mucus in the lungs, leading to frequent blockages and infections.
CF occurs due to a mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which is responsible for coding the CFTR protein (CFF 2021a).
This mutation causes the CFTR protein - which is responsible for regulating the balance of salt and water on certain body surfaces (such as the lungs) - to behave abnormally. In some cases, the CFTR protein is not produced at all (CFF 2021a).
A normal CFTR protein allows chloride (a salt component) to move from the inside of a cell to the outside, where it attracts water to the cellular surface. This water allows tiny hairs on the cellular surface (known as cilia) to sweep mucus away (CFF 2021a).
However, a defective or absent CFTR protein causes the chloride to become trapped inside the cells, preventing water from hydrating the cellular surfaces. As a result, the mucus dehydrates and becomes thicker. Furthermore, the cilia are weighed down by this thick mucus and are unable to sweep it away, causing it to accumulate (CFF 2021a).
This build-up of mucus is particularly dangerous in the lungs, as it clogs air passages and traps bacteria, leading to frequent blockages and infections. Over time, this may cause permanent lung damage (Better Health Channel 2024).
Accumulated mucus in the digestive system prevents digestive enzymes from being transferred between the pancreas and small intestine, causing issues with digesting fats and absorbing nutrients (Better Health Channel 2024).
How is Cystic Fibrosis Inherited?
CF is an autosomal recessive disease, meaning that it is not possible to inherit CF unless both parents have the defective gene (MedlinePlus 2021; Mayo Clinic 2024a).
A person who inherits only one defective gene (i.e. from only one parent) is a carrier but will not show symptoms, as the normal (unaffected gene) is dominant and will be able to compensate for the defect (Mayo Clinic 2024a).
Therefore, if both parents are carriers:
There is a 25% chance that the child will be unaffected
There is a 50% chance that the child will be a carrier
There is a 25% chance that the child will have CF.
(Mayo Clinic 2024a)
If one parent has CF and the other is a carrier:
There is a 50% chance that the child will be a carrier
There is a 50% chance that the child will have CF.
(CFF 2021c)
It is estimated that 1 in 25 Australians are carriers. While most carriers are unaware that they have the defective gene, screening services are available (Cystic Fibrosis Australia 2023).
Autosomal recessive conditions like CF can skip generations (Cystic Fibrosis Australia 2023).
Symptoms of Cystic Fibrosis
CF affects the body in a variety of ways and may cause:
A persistent cough that may produce mucus
Breathing issues
Wheezing
Frequent lung infections and sinusitis
Salty sweat
Muscle cramps or weakness in hot weather due to salt loss in sweat
Fatigue
Difficulty gaining weight
Frequent toilet visits
Bulky, greasy stools
Diarrhoea or constipation
Decreased appetite.
(Better Health Channel 2024)
Patients with CF often experience acute worsening of respiratory symptoms (known as pulmonary exacerbations (PEx)). PEx episodes often involve symptoms such as increased mucus production, increased coughing and shortness of breath along with a decrease in lung function (CFF 2021d).
Complications of Cystic Fibrosis
People with CF may experience a variety of complications, including:
Recurring lung infections
Pulmonary exacerbations (acute worsening of respiratory symptoms)
Bronchiectasis
Haemoptysis (coughing up blood due to airway damage)
Pneumothorax
Respiratory failure
Allergic bronchopulmonary aspergillosis (ABPA) (allergic reaction to fungus in the lungs)
Cardiac failure from lung damage
Cancer of the digestive tract
Malnutrition
Diabetes (affecting about 20% of teenagers with CF and up to 50% of adults with CF)
Liver disease
Kidney issues
Pancreatitis
Intestinal obstruction
Distal intestinal obstruction syndrome (DIOS)
Infertility (particularly in males)
Osteoporosis
Arthritis
Electrolyte imbalances or dehydration
Mental illness.
(Mayo Clinic 2024b; NHLBI 2024)
CF gradually damages lung tissue, decreasing lung function. In most cases, death is caused by eventual respiratory failure (NHLBI 2024; Yu et al. 2024).
Diagnosis of Cystic Fibrosis
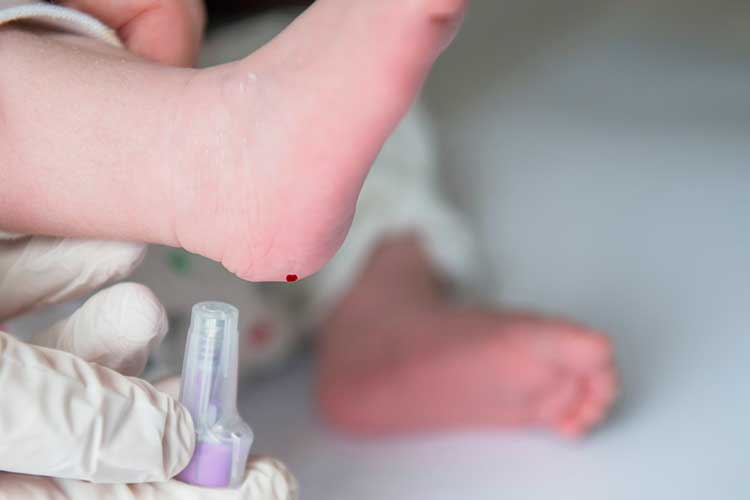
Between 48 and 72 hours after birth, neonates can undergo a heel prick test to detect CF and other medical conditions. A suspected CF diagnosis can then be confirmed by measuring the infant’s sweat for a high concentration of sodium chloride at about six weeks of age (CF Together 2024; Healthdirect 2023).
Management of Cystic Fibrosis
CF is incurable and requires intensive management to slow its progression (Better Health Channel 2024).
This may include:
Daily chest physiotherapy
A variety of medicines, which may include CFTR modulators, antibiotics and pancreatic enzyme supplements
Inhalation using a compressed air pump and nebuliser
A balanced diet that contains high amounts of protein, fat and kilojoules
 New
New